Research Interests
Professor David J Tozer

Department of Chemistry
Durham University
South Road
Durham
DH1 3LE UK
Research Interests
Electronic structure calculations are becoming an integral part of chemistry research, where they play a vital role in complementing and aiding interpretation of experimental data. To be useful, a method must provide an accurate description of the ground state of a molecule or solid, together with its response to structural, electric and magnetic perturbations. By far the most widely used electronic structure method is Kohn–Sham density functional theory (DFT), whose modest computational cost makes it applicable to large, chemically, physically and biologically relevant systems. The aim of our research is to improve the quality of DFT predictions, particularly in areas where the method is currently deficient. We collaborate with a number of international research groups, notably Helgaker (Oslo, Norway), De Proft and Geerlings (Brussels, Belgium), Ruud (Tromso, Norway), Cohen (Cambridge, UK), O’Hagan (St Andrews, UK) and Williams (Durham, UK).
Current interests include:
Exchange–Correlation Functionals
The main challenge in DFT is the accurate description of the non-classical exchange and correlation interactions between electrons. A central theme of our research is the development of improved approximations to this exchange–correlation energy. Our B97-2 approximation is a high-quality all-round functional, which has been implemented in all major commercial electronic structure programs, including Gaussian. Our KT1, KT2 and KT3 approximations were designed specifically to provide high quality NMR shielding constants and chemical shifts. Presently, we are investigating two approaces to the exchange–correlation problem. First, we are investigating Coulomb attenuated functionals, which split the Coulomb interaction into short- and long-range components. See here for further details.
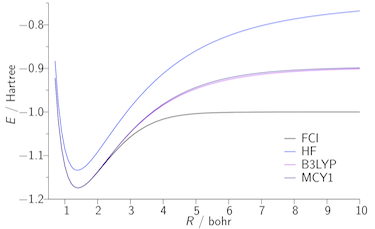
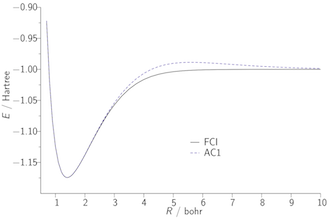
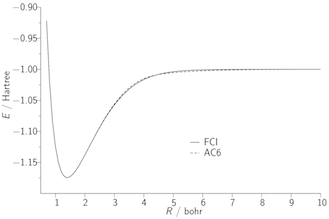
Our second approach to the exchange–correlation problem involves the adiabatic connection, which expresses the exchange–correlation energy as an integral over the electron–electron coupling strength. As an illustration, the figures below present potential energy curves of the H2 molecule; the FCI curve is exact. B3LYP and MCY are existing approximations, which fail to dissociate the molecule correctly due to the lack of static correlation. The AC1 and AC6 curves quantify what can be achieved using a simple adiabatic connection form with exact input data.
Theoretical Electronic Spectroscopy
It is well-established that DFT electronic excitation energies to charge-transfer (CT) states—for example in the dipeptide below right—are often significantly underestimated when GGA and hybrid functionals are used, whilst others such as the CT state in DMABN (left) are accurately described.
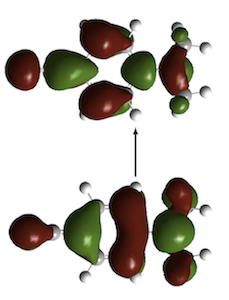
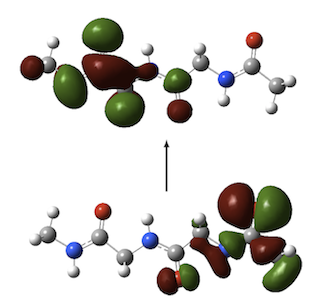
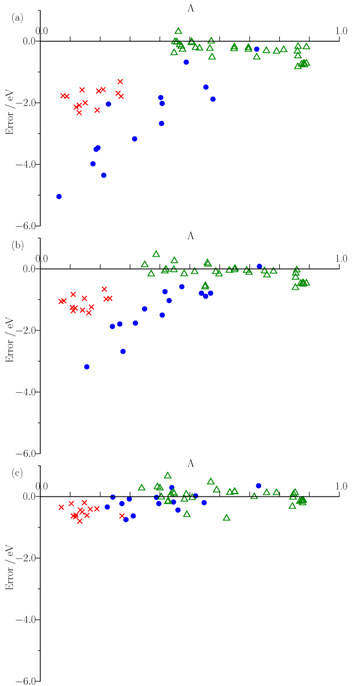
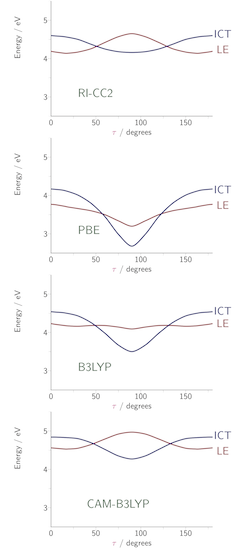
We have provided insight into this observation through a consideration of the extent to which excitation energy errors correlate with the degree of spatial overlap between the occupied and virtual orbitals involved in the excitations. The figure below plots excitation energy errors as a function of the degree of orbital overlap, Λ, for three functionals, (a) the PBE GGA, (b) the B3LYP hybrid, and (c) the CAM-B3LYP Coulomb attenuated approximation. Each point corresponds to a single excitation, with local exciations in green, Rydberg excitations in red, and CT excitations in blue. The results highlight the good quality predictions from CAM-B3LYP. For GGA and hybrid, there is a clear correlation between error and overlap, allowing a diagnostic test to be proposed. See here for full details. We have also discussed the origin of the failure in terms of the integer discontinuity in model systems.
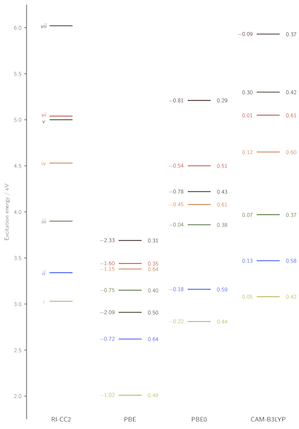
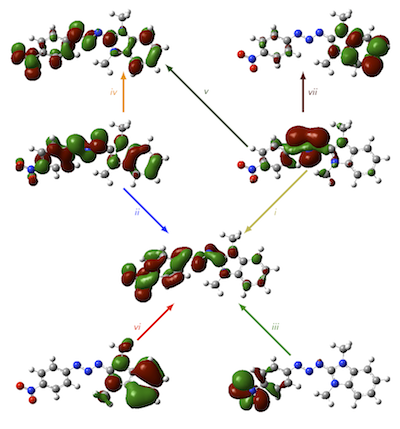
As a pertinent application of the diagonstic method and a further demonstration of the success of the CAM-B3LYP functional in describing excited states in organic molecules, we applied both to the triazene system shown below. The orbitals which represent the electronic excitations (i)-(vii) are shown, together with the excitation energy error (relative to RI-CC2) and Λ values for each transition. The success of CAM-B3LYP is evident, as is the low-overlap failure of the PBE GGA. Click the PCCP cover below for full details.

We have also applied Coulomb-attenuated functionals to the electronic (and structural) properties of polyacetylene and polyyne.
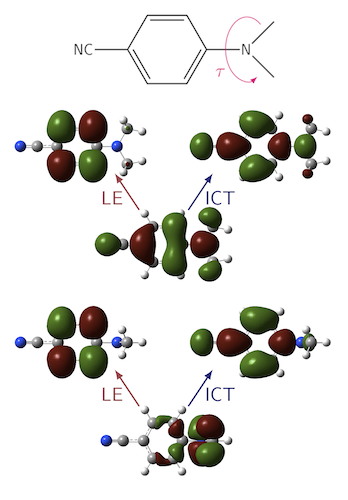
In our most recent work we have considered the implications of low overlap in excited state surfaces. For common density functional approximations, the accuracy of the surface will be non-uniform if the spatial overlap between the occupied and virtual orbitals involved in the excitation has a strong conformational dependence; the excited state surface will collapse toward the ground state in regions where the overlap is very low. This is illustrated below for DMABN. For both the LE and ICT excitations, the overlap drops very low upon twisting and so both states collapse with PBE and to a lesser extent B3LYP. This breakdown is eliminated using CAM-B3LYP. See here for full details.
Magnetic Response Parameters; Kohn–Sham Equations; Optimised Effective Potentials
For hybrid functionals, we have demonstrated that the quality of magnetic response parameters is highly sensitive to the formulation of the Kohn–Sham equations – significant improvements are obtained when the equations involve a well-defined multiplicative potential. Our investigations have considered the Wu–Yang/Zhao–Morrison–Parr approaches, together with the localised Hartree–Fock approximation (LHF) and the rigorous optimised effective potential approach (OEP); we have highlighted unexpected difference between the latter two methods. The figures below compare the LHF and OEP potentials and electron densities. See here for further details.
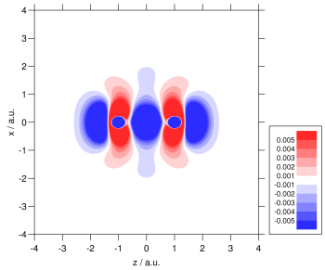
Recent studies have focused on transition metal NMR chemical shifts and rotational g tensors; the correlation plot below highlights the accuracy in the latter. Related properties of interest include indirect nuclear spin–spin coupling constants and NMR of the solid state.
Conceptual DFT; Studies of Temporary Anions
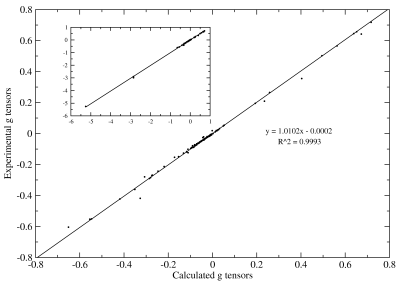
Conceptual DFT is concerned with the calculation of familiar chemical concepts such as hardness, softness, and electronegativity, from quantities such as the ionisation potential and electron affinity. We have developed a novel method for calculating negative electron affinities, which avoids an explicit calculation on the (problematic) temporary anion. The method yields results that correlate well with experimental values from electron transmission spectroscopy. Insight into the method is provided by an analysis of the integer discontinuity. We have recently extended this work to allow explicit DFT calculations on temporary anions. The left hand figure below shows the singly occupied molecular orbital (left) and spin density (right) in the adenine anion. Part (a) is a conventional DFT calculation with a diffuse basis set—the electron is clearly leaving the system. Parts (b) and (c) use our proposed approach with a potential wall and optimally compact set, respectively. Click on the PCCP cover below for full details.

Other recent work includes an investigation of confinement effects on chemical reactivity.
Insight from High Quality Electron Densities
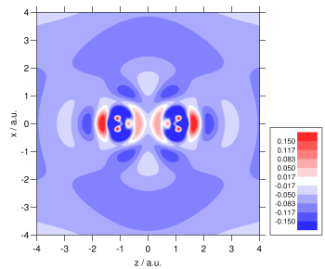
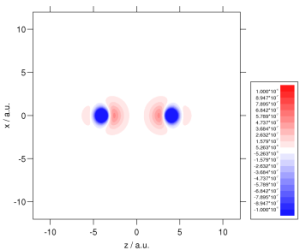
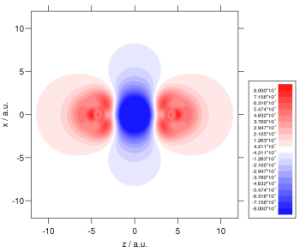
We make extensive use of high quality ab initio electron densities, in order to help us understand why DFT sometimes fails and hence improve the theory. For example, investigations demonstrated that an improved electron density could reduce DFT shielding constant errors by almost a factor of three. Ab initio densities also help us to understand why DFT fails to describe long-range der Waals interactions– it fails to distort the electron densities towards one another. The plots below show the density distortion and associated interaction correlation potential in the helium dimer, determined using coupled cluster theory; local DFT functionals cannot reproduce this behaviour! See here for further details. It is vital to resolve this deficiency if the full potential of DFT is to be realised in the field of intermolecular forces. We are also using high quality densities in our investigations of the adiabatic connection (see above).
Potential Energy Surfaces and Conformational Preferences
In addition to theory development, we apply DFT to real chemical and biological problems. We are particularly interested in the calculation of theoretically challenging potential energy surfaces (e.g. oxirene-ketene interconversion) and the study of conformational preferences in organofluorine chemistry, arising due to the gauche effect or intramolecular electrostatic interactions. Click on the figure below for another example.